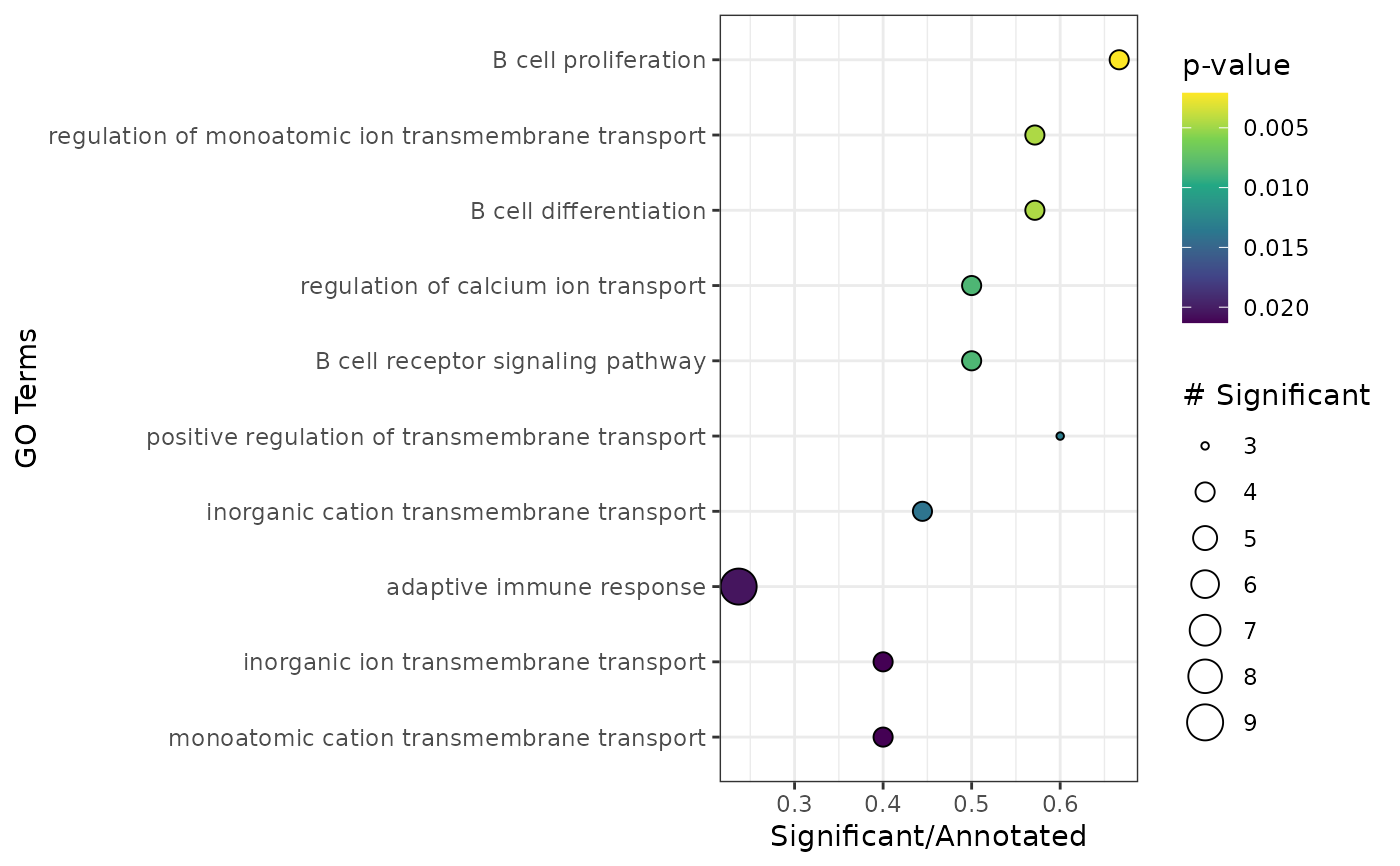
Run Gene overrepresentation analysis with topGO
apl_topGO.RdThis function uses the Kolmogorov-Smirnov test as implemented by the package topGO to test for overrepresentation in Gene Ontology gene sets.
Usage
apl_topGO(
caobj,
ontology,
organism = "hs",
ngenes = 1000,
score_cutoff = 0,
use_coords = FALSE,
return_plot = FALSE,
top_res = 15
)Arguments
- caobj
A "cacomp" object with principal row coordinates and standardized column coordinates calculated.
- ontology
Character string. Chooses GO sets for 'BP' (biological processes), 'CC' (cell compartment) or 'MF' (molecular function).
- organism
Character string. Either 'hs' (homo sapiens), 'mm' (mus musculus) or the name of the organism package such as 'org.*.eg.db'.
- ngenes
Numeric. Number of top ranked genes to test for overrepresentation.
- score_cutoff
numeric. S-alpha score cutoff. Only genes with a score larger will be tested.
- use_coords
Logical. Whether the x-coordinates of the row APL coordinates should be used for ranking. Only recommended when no S-alpha score (see apl_score()) can be calculated.
- return_plot
Logical. Whether a plot of significant gene sets should be additionally returned.
- top_res
Numeric. Number of top scoring genes to plot.
Details
For a chosen group of cells/samples, the top 'ngenes' group specific genes are used for gene overrepresentation analysis. The genes are ranked either by the precomputed APL score, or, if not available by their APL x-coordinates.
References
Adrian Alexa and Jorg Rahnenfuhrer
topGO: Enrichment Analysis for Gene Ontology.
R package version 2.42.0.
Examples
library(SeuratObject)
#> Loading required package: sp
#> ‘SeuratObject’ was built under R 4.4.0 but the current version is
#> 4.4.2; it is recomended that you reinstall ‘SeuratObject’ as the ABI
#> for R may have changed
#>
#> Attaching package: ‘SeuratObject’
#> The following objects are masked from ‘package:base’:
#>
#> intersect, t
set.seed(1234)
cnts <- SeuratObject::LayerData(pbmc_small, assay = "RNA", layer = "counts")
cnts <- as.matrix(cnts)
# Run CA on example from Seurat
ca <- cacomp(pbmc_small,
princ_coords = 3,
return_input = FALSE,
assay = "RNA",
slot = "counts")
#> Warning:
#> Parameter top is >nrow(obj) and therefore ignored.
#> No dimensions specified. Setting dimensions to: 15
grp <- which(Idents(pbmc_small) == 2)
ca <- apl_coords(ca, group = grp)
ca <- apl_score(ca,
mat = cnts)
#>
|
| | 0%
|
|======= | 10%
|
|============== | 20%
|
|===================== | 30%
|
|============================ | 40%
|
|=================================== | 50%
|
|========================================== | 60%
|
|================================================= | 70%
|
|======================================================== | 80%
|
|=============================================================== | 90%
|
|======================================================================| 100%
enr <- apl_topGO(ca,
ontology = "BP",
organism = "hs")
#>
#> groupGOTerms: GOBPTerm, GOMFTerm, GOCCTerm environments built.
#> Loading required package: org.Hs.eg.db
#> Loading required package: AnnotationDbi
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#>
#> Attaching package: ‘BiocGenerics’
#> The following object is masked from ‘package:SeuratObject’:
#>
#> intersect
#> The following objects are masked from ‘package:stats’:
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from ‘package:base’:
#>
#> Filter, Find, Map, Position, Reduce, anyDuplicated, aperm, append,
#> as.data.frame, basename, cbind, colnames, dirname, do.call,
#> duplicated, eval, evalq, get, grep, grepl, intersect, is.unsorted,
#> lapply, mapply, match, mget, order, paste, pmax, pmax.int, pmin,
#> pmin.int, rank, rbind, rownames, sapply, saveRDS, setdiff, table,
#> tapply, union, unique, unsplit, which.max, which.min
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#> Loading required package: IRanges
#> Loading required package: S4Vectors
#>
#> Attaching package: ‘S4Vectors’
#> The following object is masked from ‘package:utils’:
#>
#> findMatches
#> The following objects are masked from ‘package:base’:
#>
#> I, expand.grid, unname
#>
#> Attaching package: ‘IRanges’
#> The following object is masked from ‘package:sp’:
#>
#> %over%
#>
#> Building most specific GOs .....
#> ( 1348 GO terms found. )
#>
#> Build GO DAG topology ..........
#> ( 3594 GO terms and 7817 relations. )
#>
#> Annotating nodes ...............
#> ( 207 genes annotated to the GO terms. )
#>
#> -- Elim Algorithm --
#>
#> the algorithm is scoring 519 nontrivial nodes
#> parameters:
#> test statistic: fisher
#> cutOff: 0.01
#>
#> Level 12: 1 nodes to be scored (0 eliminated genes)
#>
#> Level 11: 7 nodes to be scored (0 eliminated genes)
#>
#> Level 10: 17 nodes to be scored (8 eliminated genes)
#>
#> Level 9: 24 nodes to be scored (11 eliminated genes)
#>
#> Level 8: 53 nodes to be scored (17 eliminated genes)
#>
#> Level 7: 74 nodes to be scored (19 eliminated genes)
#>
#> Level 6: 103 nodes to be scored (27 eliminated genes)
#>
#> Level 5: 101 nodes to be scored (27 eliminated genes)
#>
#> Level 4: 74 nodes to be scored (27 eliminated genes)
#>
#> Level 3: 50 nodes to be scored (27 eliminated genes)
#>
#> Level 2: 14 nodes to be scored (27 eliminated genes)
#>
#> Level 1: 1 nodes to be scored (27 eliminated genes)
plot_enrichment(enr)